
Back to top
Genotyping-by-Sequencing (GBS)

With the rapid progress of next generation sequencing technologies, Genotyping-by-Sequencing became available for a very attractive price.
In contrast to more traditional genotyping methods, SNP (Single Nucleotide Polymorphism) genotyping by next generation sequencing does not require previous marker development or sequence information but allows direct SNP genotyping within any population of interest.
We are happy to assist your project with this breakthrough technology, please don’t hesitate to contact us to discuss possible project layouts.
Workflow
A typical workflow for a Genotyping-by-Sequencing project is shown in the figure below. Please note that our modular genotyping process allows various entry and output options. Whether you wish to outsource the entire Genotyping-by-Sequencing project or only parts of it, is up to you to decide.
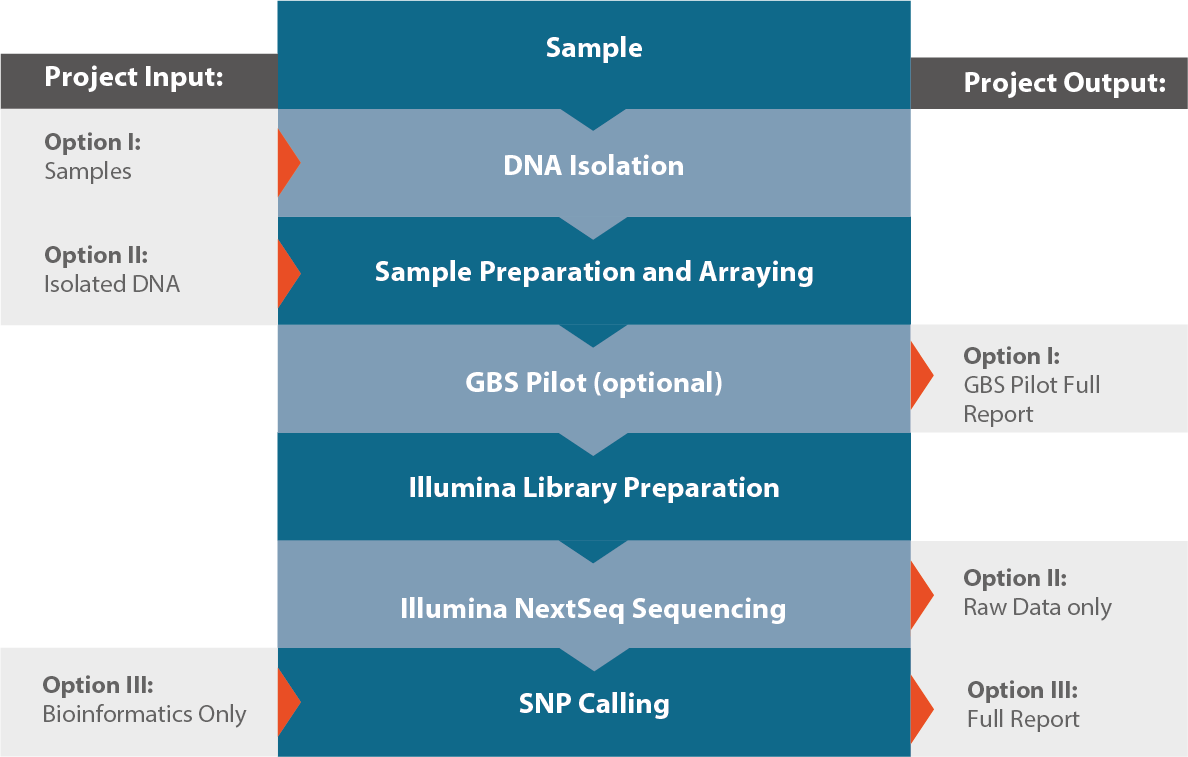
Product
GBS Pilot
With the GBS Pilot we offer the possibility to test and optimize the GBS protocol for your species on a small sample subset (5-10 samples). This service includes the preparation of individually MID-tagged reduced representation libraries, the sequencing on the Illumina MiSeq platform and the bioinformatic analysis. This option is especially advantageous for medium sized projects or for method validation purposes.
GBS Batch Run
This service includes the preparation of individually MID-tagged reduced representation libraries and the sequencing on an Illumina NextSeq platform. Depending on your goals we offer the sequencing of different batch sizes as well as sequencing read lengths. The GBS Batch Runs makes sense in case larger batches of samples can be analyzed in parallel.
Bioinformatics
As the analysis of large and complex GBS datasets requires some specific knowledge in bioinformatics, we can run the basic analysis to identify SNPs in the dataset. The analysis will include the clustering of highly similar sequences for each sample (i.e. stacking), the identification of SNPs within samples as well as identifying SNP loci across the samples included in the study (i.e. catalogue construction). The results will be a table including the SNP call per locus and sample.
In case that a reference genome is available for your species, the stack sequence cluster will be mapped on the genome allowing you to get additional information on each SNPs' location.
Our standard output file is the VCF (Variant Call Format). In case you need a specific file format for further analysis, contact us and we will find a solution for you.
Results
The delivery of a Genotyping-by-Sequencing project includes:
- a GBS project summary
- the raw data (fastq files)
In case we perform the sequence assembly and filtering for SNP candidates with the STACKS package, you will additionally receive a table containing the SNP call per locus and sample (Figure 1). Further, if provided, the information of a reference genome will be included in the analysis (Figure 2).
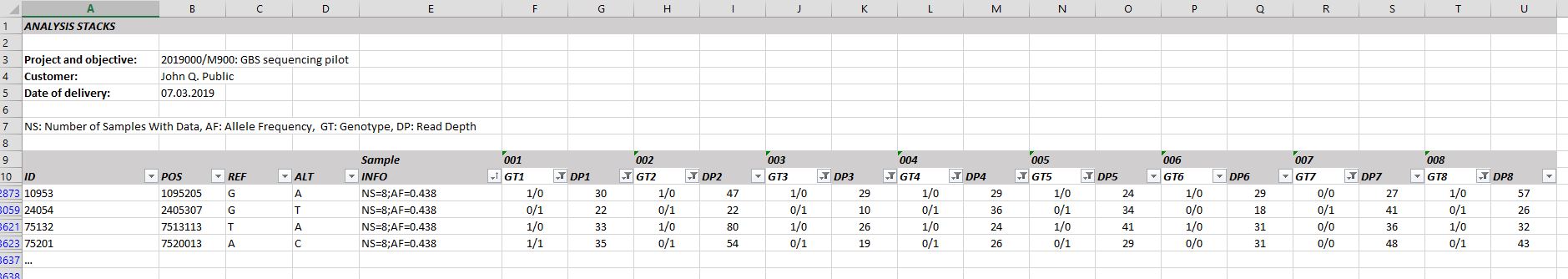
Figure 1. Genotype table including SNP calls per locus and sample for a reference genome-based analysis strategy.

Figure 2. Schematic representation of GBS data analysis strategies. In case a reference genome is available, the sequence reads derived from the reduced representation libraries are mapped against the reference genome and then the SNPs are integrated resulting in a genotype table incl. the coordinate of the SNP on the reference genome. This information is especially helpful if PCR-based assays shall be developed for a limited set of SNPs. In contrast, a de-novo analysis strategy can be applied for any non-model organisms resulting in a genotype table. However, in the de-novo analysis strategy no information is available on the locations of the SNPs.